07 Virtual Screening
Author
Joshua Schrier
Title
07 Virtual Screening
Description
Perform virtual screening against PubChem using ligand-based approach.
Category
Educational Materials
Keywords
cheminformatics, Chemoinformatics, chemical information, PubChem, quantitative structure, property relationships, QSPR, machine learning, computer-aided drug design, chemistry
URL
http://www.notebookarchive.org/2020-10-ebo06m8/
DOI
https://notebookarchive.org/2020-10-ebo06m8
Date Added
2020-10-31
Date Last Modified
2020-10-31
File Size
2.24 megabytes
Supplements
Rights
CC BY-NC-SA 4.0

Virtual Screening
Virtual Screening
Objectives
Objectives
◼
Perform virtual screening against PubChem using ligand-based approach.
◼
Apply filters to prioritize virtual screening hit list.
◼
Manipulate Dataset representations.
In this notebook, we perform virtual screening against PubChem using a set of known ligands for muscle glycogen phosphorylase. Compound filters will be applied to identify drug-like compounds and unique structures in terms of canonical SMILES will be selected to remove redundant structures. For some top-ranked compounds in the list, their binding mode will be predicted using molecular docking (which will be covered in a separate assignment).
We will be making extensive use of the Dataset functionality in Mathematica. Please take a few minutes to review the documentation. Another useful resource is Seth Chandler’s “Primer on Associations and Datasets:
https://community.wolfram.com/groups/-/m/t/1167544
We will be making extensive use of the Dataset functionality in Mathematica. Please take a few minutes to review the documentation. Another useful resource is Seth Chandler’s “Primer on Associations and Datasets:
https://community.wolfram.com/groups/-/m/t/1167544
Read known ligands from a file.
Read known ligands from a file.
As a starting point, let’s download a set of known ligands against muscle glycogen phosphorylase. These data are obtained from the DUD-E (Directory of Useful Decoys, Enhanced) data sets (http://dude.docking.org/), which contain known actives and inactives for 102 protein targets. The DUD-E sets are widely used in benchmarking studies that compare the performance of different virtual screening approaches (https://doi.org/10.1021/jm300687e).
Go to the DUD-E target page (http://dude.docking.org/targets) and find muscle glycogen phosphorylase (Target Name: PYGM, PDB ID: 1c8k) from the target list. Clicking the target name “PYGM” directs you to the page that lists various files (http://dude.docking.org/targets/pygm). We are interested in the file “actives_final.ism”, which contains the SMILES strings of known actives, and will download this directly by using the file’s URL. However, you are encouraged to download and open this file in a text editor (e.g. TextEdit or WordPad) to check the format of this file.
For convenience we will use the DatasetWithHeaders repository function to add the necessary headers to the file. (This could be avoided by adding column names to the downloaded copy of the file before Import-ing the file.) Here it goes:
Go to the DUD-E target page (http://dude.docking.org/targets) and find muscle glycogen phosphorylase (Target Name: PYGM, PDB ID: 1c8k) from the target list. Clicking the target name “PYGM” directs you to the page that lists various files (http://dude.docking.org/targets/pygm). We are interested in the file “actives_final.ism”, which contains the SMILES strings of known actives, and will download this directly by using the file’s URL. However, you are encouraged to download and open this file in a text editor (e.g. TextEdit or WordPad) to check the format of this file.
For convenience we will use the DatasetWithHeaders repository function to add the necessary headers to the file. (This could be avoided by adding column names to the downloaded copy of the file before Import-ing the file.) Here it goes:
In[]:=
dfAct=ResourceFunction["DatasetWithHeaders"][Import["http://dude.docking.org/targets/pygm/actives_final.ism"],(*filetoimport*){"smiles","dat","id"}(*headernamestoadd*)]
Out[]=
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Similarity Search against PubChem
Similarity Search against PubChem
Now, let’s perform similarity search against PubChem using each known active compound as a query. There are a few things to mention in this step:
◼
The isomeric SMILES string is available for each query compound. This string will be used to specify the input structure, so HTTP POST should be used. (Please review Assignment 2A on the difference between GET and POST.)
◼
Unlike the examples in Assignment 6, all similarity comparisons are performed on the PubChem server.
◼
During PubChem’s similarity search, molecular similarity is evaluated using the PubChem fingerprints and Tanimoto coefficient. By default, similarity search will return compounds with Tanimoto scores of 0.9 or higher. While we will use the default threshold in this practice, it is noteworthy that it is adjustable. If you use a higher threshold (e.g., 0.99), you will get a fewer hits, which are too similar to the query compounds. If you use a lower threshold (e.g., 0.88), you will get more hits, but they will include more false positives.
◼
PubChem’s similarity search does not return the similarity scores between the query and hit compounds. Only the hit compound list is returned, which makes it difficult to rank the hit compounds for compound selection. To address this issue, for each hit compound, we compute the number of query compounds that returned that compound as a hit. [Because we are using multiple query compounds for similarity search, it is possible for different query compounds to return the same compound as a hit. That is, the hit compound may be similar to multiple query compounds. The underlying assumption is that hit compounds returned multiple times from different queries are more likely to be active than those returned only once from a single query.]
◼
Important Reminder: Because new compounds are added to PubChem regularly, your results may not exactly match the results archived here (October 2020). You should have more compounds returned, and the number of similar matches to each of the query compounds may have increased. So keep in mind that if you get different results, you are not necessarily doing anything wrong.
Using PUG-REST
Using PUG-REST
In[]:=
similaritySearch[SMILES_String]:=With[{url=URLBuild[{"https://pubchem.ncbi.nlm.nih.gov/rest/pug","compound/fastsimilarity_2d/smiles/cids/txt"}]},URLExecute[#,"CSV"]&@HTTPRequest[url,<|"Method""POST","Body"{"smiles"SMILES}|>]//Flatten]
Define a list of SMILES strings:
In[]:=
smilesAct=dfAct[All,"smiles"]//Normal;
Test it on a single SMILES string input:
In[]:=
similaritySearch@First@smilesAct
Out[]=
{44354844,44354843,44354829,44354455,44354454,44354446,44354391,44354362,44354301,44354099}
Having confirmed that it works, then go and apply it to each of the SMILES strings in our compound list (this will take a few minutes, including the pauses), using and AssociationMap over the list of SMILES strings:
In[]:=
cidsHitsForEachExample=AssociationMap[Pause[0.2];similaritySearch[#]&,smilesAct];
In[]:=
Length@Flatten@Values@cidsHitsForEachExample
Out[]=
67613
However, many of these are duplicates. Let us combine them, and form a new Association whose keys are the CID and whose values are the Counts of how many times that occurred. We then ReverseSort (by the association’s value) to rank the most common matches:
In[]:=
cidsHits=ReverseSort@Counts@Flatten@Values@cidsHitsForEachExample
Out[]=
4435434816,4435432215,4435434915,4435437015,4435790713,1177985412,2197459512,4435793712,4435793812,4435435012,4435430112,4435436212,4435445412,4435445512, ⋯25949⋯ | |||||
|
How many total hits were found? (as of October 2020):
In[]:=
Length[cidsHits]
Out[]=
25976
The vast majority of cases only have a single hit:
In[]:=
Histogram[cidsHits]
Out[]=

Using PubChem Service Connection
Using PubChem Service Connection
We can perform the same search using the PubChem Service Connection; the Method option (described under “Parameter Details” allows us to define a similarity method. As a first example, let us look at the similarity of the first entry in smilesAct:
In[]:=
smilesAct=dfAct[All,"smiles"]//Normal;ServiceExecute["PubChem","CompoundCID",{"SMILES"First@smilesAct,Method->"Similarity2DSearch"}]
Out[]=
|
It will be more convenient to merge the results together if the results are in the form of a List instead of a Dataset. One way to solve this is by defining a function that converts the result into an ordinary list. The final result will be identical to the the “similaritySearch” function defined above:
In[]:=
similaritySearch[SMILES_String]:=ServiceExecute["PubChem","CompoundCID",{"SMILES"SMILES,Method->"Similarity2DSearch"}]//Values//Normal
In[]:=
similaritySearch[First@smilesAct](*demo*)
Out[]=
{{44354844,44354843,44354829,44354455,44354454,44354446,44354391,44354362,44354301,44354099}}
This function can be substituted for the “similaritySearch” function above, to achieve all of the subsequent results.
Exclude the query compounds from the hits
Exclude the query compounds from the hits
Using PUG-REST
Using PUG-REST
In the previous step, we repeated similarity searches using multiple query molecules. This may result in a query molecule being returned as a hit from similarity search using another query molecule. Therefore, we want to check if the hit compound list has any query compounds and if any, we want to remove them. Below, we search PubChem for compounds identical to the query molecules and remove them from the hit compound list.
Note that the identity_type parameter in the PUG-REST request is set to “same_connectivity”, which will return compounds with the same connectivity with the query molecule (ignoring stereochemistry and isotope information). The default for this parameter is “same_stereo_isotope”, which returns compounds with the same stereochemistry AND isotope information.
Note that the identity_type parameter in the PUG-REST request is set to “same_connectivity”, which will return compounds with the same connectivity with the query molecule (ignoring stereochemistry and isotope information). The default for this parameter is “same_stereo_isotope”, which returns compounds with the same stereochemistry AND isotope information.
In[]:=
identitySearch[SMILES_String]:=With[{url=URLBuild[{"https://pubchem.ncbi.nlm.nih.gov/rest/pug","compound/fastidentity/smiles/cids/txt"},{"identity_type""same_connectivity"}]},URLExecute[#,"CSV"]&@HTTPRequest[url,<|"Method""POST","Body"{"smiles"SMILES}|>]//Flatten]
In[]:=
cidsQuery=AssociationMap[Pause[0.2];identitySearch[#]&,smilesAct];
In[]:=
queryCompounds=Flatten@Values@cidsQuery;Length@queryCompounds(*ShowthenumberofCIDsthatrepresentthequerycompounds.*)
Out[]=
138
Now remove the query compounds from the cidsHits list by dropping the Keys that are contained in the queryCompounds list. The KeyDropFrom function modifies the association “in place” (in contrast to the KeyDrop function that returns a new association):
In[]:=
KeyDropFrom[cidsHits,queryCompounds]
Out[]=
1177985412,9307706511,9307706411,5180871811,4536969611,1760071611,5301334911,14113591511,11807885811,12956752410,12956752310,12956751410,12956751310, ⋯25812⋯ | |||||
|
In comparison to the previous result, many of the most common results have been eliminated.
Using the PubChem Service Connection
Using the PubChem Service Connection
This type of query can also be performed using the PubChem Service Connection. The wrapper function converts the returned Dataset into a Normal list, so that the results are identical to the previous definition of the function:
In[]:=
identitySearch[SMILES_String]:=ServiceExecute["PubChem","CompoundCID",{"SMILES"SMILES,Method->"SameConnectivity"}]//Values//Normal
Filtering out non-drug-like compounds
Filtering out non-drug-like compounds
Retrieving the data
Retrieving the data
In this step, non-drug-like compounds are filtered out from the list. To do that, four molecular properties are downloaded from PubChem and stored in a Dataset. It is probably easiest to do this using the PubChem service. Furthermore, because this might take a few minutes, we’ll use the DynamicMap repository function to show a progress indicator while the queries are taking place:
In[]:=
requestProperties[cids_List,chunkSize_Integer:100,properties_List:{"HBondDonorCount","HBondAcceptorCount","MolecularWeight","XLogP","CanonicalSMILES","IsomericSMILES"}]:=With[{cidsBatches=Partition[cids,UpTo[chunkSize]]},Join@@ResourceFunction["DynamicMap"][Pause[0.2];ServiceExecute["PubChem","CompoundProperties",{"CompoundID"#,"Property"properties}]&,cidsBatches]]
Having defined the function, apply it to the list of CIDs. This will take a few minutes:
In[]:=
dfRaw=requestProperties@Keys@cidsHits
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Notice how the molecular weight is parsed as a unit-bearing Quantity. This can be useful, as it facilitates dimensional calculations. On the other hand, dimensioned quantities can get in the way of some of the linear fitting and machine learning tasks that we might perform with this dataset. Furthermore, other programs may not be able to work with the dimensioned quantities. For simplicity, let’s remove convert these into ordinary (non-dimensioned) numbers by selectively applying the QuantityMagnitude function to the fourth column, leaving the others unchanged; we’ll save the result back into the same variable:
In[]:=
dfRaw=dfRaw[All,{4->QuantityMagnitude}]
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Reminder: Do not be alarmed if you have more than this number of entries when you run the calculations. Recall that new compounds are added to PubChem every day, and so there are potentially more molecules that will have been returned by the similarity search in the first part of this exercise.
Storing and retrieving variable definitions for resuming lengthy calculations
Storing and retrieving variable definitions for resuming lengthy calculations
Let’s save the results as a CSV file for later use—this way you can restart your investigation without having to wait for the query a second time. We can either save the variable itself into a file (“hits.wl”) using Put:
In[]:=
Put[dfRaw,"hits.wl"]
Put also has the abbreviation “>>”; the expression below is identical to the expression above:
In[]:=
dfRaw>>"hits.wl"
Alternatively we can Export the contents of the variable into a format that can be used by other programs, such as comma-separated value (CSV) format:
In[]:=
Export["hits.csv",dfRaw]
Out[]=
hits.csv
Should we want to restart the analysis, we do not need to perform the query again. Instead, we can resume by reading the stored results. Variables saved using Put can be retrieved using Get:
In[]:=
Get["hits.wl"]
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This results in the definition of the saved variable (dfRaw) with the same value. It is now in memory and can be used as if we had defined it by any other process. Like Put, Get has an abbreviation “<<”; the expression below is identical to the expression above:
In[]:=
<<"hits.wl";
Alternatively, files Export-ed to CSV or other text-based formats can be Import-ed, as we have done above. Saving these intermediate results allows us to restart our work (starting from the “Get”) without having to reload the data. This is especially advantageous for lengthy calculations.
When should one prefer Get/Put to Import/Export? In general, for data consisting of ordinary numerical and string datatypes, there is very little difference. An advantage to Get/Put is that it also allows us to save more complicated data types (e.g., Molecule, images, etc.). An advantage of Import/Export is that it allows us to use the saved file in other programs besides Mathematica.
When should one prefer Get/Put to Import/Export? In general, for data consisting of ordinary numerical and string datatypes, there is very little difference. An advantage to Get/Put is that it also allows us to save more complicated data types (e.g., Molecule, images, etc.). An advantage of Import/Export is that it allows us to use the saved file in other programs besides Mathematica.
Data Handling: Dealing with Missing Entries and Joining Datasets
Data Handling: Dealing with Missing Entries and Joining Datasets
Take a few seconds to explore the Dataset browser (by removing the semicolon from the above Import line), and observe that some compounds do not have computed XLogP values (because the XLogP algorithm cannot handle inorganic compounds, salts, and mixtures) and we want to remove them. One can manually apply a function (e.g., “Count[_Missing]”) to a specifically named column (e.g., “XLogP”):
In[]:=
dfRaw[Count[_Missing],"XLogP"]
Out[]=
752
A more elegant solution to apply a function to all columns is to first Transpose the dataset, and then apply the function to each of the resulting rows:
In[]:=
dfRaw[Transpose][All,Count[_Missing]]
Out[]=
|
Now, we would also like to incorporate the data stored in cidHits; this was constructed as an Association with keys as the CompoundID and values of the number of matches returned. One approach is to construct a Dataset by performing a KeyValueMap to create a list of associations with columns name matching the Dataset constructed above:
In[]:=
dfFreq=KeyValueMap[<|"CompoundID"#1,"HitFreq"#2|>&,cidsHits]//Dataset
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||
We can join the two sets of data on the common values of “CompoundID” column using the JoinAcross function. The result is saved into a new Dataset called “df”. You may need to scroll the browser to see that the “HitFreq” column has been added to the far right:
In[]:=
df=JoinAcross[dfRaw,dfFreq,"CompoundID"]Dimensions[df]
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Out[]=
{25838,8}
Now, let’s sort by “HitFreq”. For column names that involve only letters and numbers, “#ColumnName &” is a valid description. In this case, we want to reverse sort each column by the value of the column “HitFreq”:
In[]:=
df[ReverseSortBy[#HitFreq&]]
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For more complicated column names, such as those with hyphens or other characters, the string name of the column can be used as shown below:
In[]:=
df[ReverseSortBy[#["HitFreq"]&]]
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Constructing Queries
Constructing Queries
Now identify and remove those compounds that satisfy all criteria of Lipinski's rule of five. There are two ways to do this. The first is to apply a function directly to the Dataset. For example, one part of the rule of five is that the number of hydrogen bond donors is less-than-or-equal-to five:
In[]:=
df[Select[#HBondDonorCount≤5&]]//Length
Out[]=
25775
In[]:=
q=Query[Select[(#HBondDonorCount≤5)&]](*definethequery*)q[df]//Length(*applythequerytodf,andcountthenumberofentries*)
Out[]=
Query[Select[#HBondDonorCount≤5&]]
Out[]=
25775
In[]:=
lipinski=Query[Select[(#HBondDonorCount≤5)&&(#HBondAcceptorCount≤10)&&(#MolecularWeight≤500)&&(#XLogP≤5)&]];
And then applied to the Dataset to remove non-druglike molecules, substantially reducing the number of candidate molecules:
In[]:=
lipinski@df
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Draw the structures of the top 10 compounds
Draw the structures of the top 10 compounds
Let’s check the structure of the top 10 compounds in the hit list. To do this, we’ll define a Query for the top-n entries:
In[]:=
top[n_Integer]:=Query[TakeLargestBy[#HitFreq&,n],{"CompoundID","IsomericSMILES"}]
Observe that this function returns a Query that can be applied to the Dataset. Queries can be applied sequentially; it might be convenient to first apply the Lipinski rule-of-five Query defined above and then use this to take the top entries. If these were more computationally intensive queries, we would save the intermediate results, but in the present case these are inexpensive enough to recompute as needed:
In[]:=
cidsTop=top[10]@lipinski@df
Out[]=
|
(Again, this is the periodic reminder that new molecules are added to PubChem daily, and so the CompoundIDs in your top-10 list may be different from the one above, generated in October 2020.)
Next, we define a Query that generates the molecule image and apply it to our Dataset:
Next, we define a Query that generates the molecule image and apply it to our Dataset:
In[]:=
compoundImages=Query[All,<|"CompoundID"#CompoundID,"Image"->MoleculePlot@Molecule@#IsomericSMILES|>&]
Out[]=
Query[All,Association[CompoundID#CompoundID,ImageMoleculePlot[Molecule[#IsomericSMILES]]]&]
In[]:=
compoundImages@top[10]@lipinski@df
Out[]=
|










An important observation is that the hit list contains multiple compounds with the same connectivity. For example, CIDs 93077065 and 93077064 are stereoisomers of each other and CID 53013349 has the same connectivity as both of those two CIDs, but its stereocenter is unspecified. When performing a screening with limited resources in the early stage of drug discovery, you may want to test as diverse molecules as possible, avoiding testing too similar structures.
To do so, let’s look into PubChem’s canonical SMILES strings, which do not encode the stereochemistry and isotope information. Chemicals with the same connectivity but with different stereochemistry/isotopes should have the same canonical SMILES. In the next section, we select unique compounds in terms of canonical SMILES to reduce the number of compounds to screen.
To do so, let’s look into PubChem’s canonical SMILES strings, which do not encode the stereochemistry and isotope information. Chemicals with the same connectivity but with different stereochemistry/isotopes should have the same canonical SMILES. In the next section, we select unique compounds in terms of canonical SMILES to reduce the number of compounds to screen.
Extract unique compounds in terms of canonical SMILES
Extract unique compounds in terms of canonical SMILES
The next few cells show how to get unique values within a column (in this case, unique canonical SMILES). We will demonstrate how to build this up sequentially. First, we want to group the entries by the “CanonicalSMILES” entry; this creates a set of groups (e.g., observe how some have multiple entries):
In[]:=
df[DeleteDuplicatesBy["CanonicalSMILES"]]
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In[]:=
uniqueCanonicalSMILES=Query[DeleteDuplicatesBy["CanonicalSMILES"]];
and then apply this as part of a pipeline of queries:
In[]:=
pygmDF=lipinski@uniqueCanonicalSMILES@df
Out[]=
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
And the resulting dataset can be queries to generate images as before:
In[]:=
compoundImages@top[10]@pygmDF
Out[]=
|







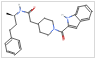

Generating structures and saving molecules in files
Generating structures and saving molecules in files
Now save the molecules in the cids_top list in files, which will be used in molecular docking experiments. For simplicity, we will use only the top three compounds in the list:
In[]:=
cidsTop=top[3]@pygmDF
Out[]=
|
The MoleculeModify performs transformations on Molecule objects; here we want to minimize the MMFF energy. Before applying it to the the lead molecules, let’s briefly explore how it works by demonstrating on ethanol:
In[]:=
MoleculeModify["EnergyMinimizeAtomCoordinates"]@Molecule["CCO"]
Out[]=
Molecule
 |  |
|
What does the resulting Molecule look like?
In[]:=
MoleculePlot3D[%]
Out[]=

We can use MoleculeModify to define a Query to add the optimized structures to the Dataset:
In[]:=
addStructures=Query[All,<|"CompoundID"#CompoundID,"structure"->MoleculeModify["EnergyMinimizeAtomCoordinates"]@Molecule[#IsomericSMILES]|>&];addStructures@cidsTop
Out[]=
|
The optimized structures can be Export-ed in a variety of formats, that can be read by docking software:
In[]:=
Map[Export["pygm_lig_"<>ToString[#CompoundID]<>".mol",#structure]&,addStructures@cidsTop]
Out[]=
|
Finally, let’s save all the compound information for later use:
In[]:=
Export["pygm_df.csv",pygmDF]
Out[]=
pygm_df.csv
Exercises
Exercises
Exercise 1. From the DUD-E target list (http://dude.docking.org/targets), find cyclooxigenase-2 (Target Name: PGH2, PDB ID: 3ln1). Download the “actives_final.ism” file and save it as “pgh2_3ln1_actives.ism” or copy the file’s URL. Load the data into a data frame called df_act. After loading the data, show the following information:
◼
the number of rows of the data frame.
◼
the first five rows of the data frame.
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 2. Perform similarity search using each of the isomeric SMILES contained in the loaded data frame.
◼
As we did for PYGM ligands in this notebook, track the number of times a particular hit is returned from multiple queries, using an association named cidsHit (CIDs as keys and the frequencies as values). This information will be used to rank the hit compounds.
◼
Print the total number of hits returned from this step (which is the same as the number of CIDs in cids_hit.
◼
Add Pause[0.2] to avoid overload the PubChem servers.
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 3. The hit list from the above step may contain the query compounds themselves. Get the CIDs of the query compounds through identity search and remove them from the hit list.
◼
Set the optional parameter “identity_type” to “same_connectivity”.
◼
◼
Print the number of CIDs correspond to the query compounds.
◼
Print the number of the remaining hit compounds, after removing the query compounds from the hit list.
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 4. Download the hydrogen donor and acceptor counts, molecular weights, XlogP, number of rotatable bonds, and canonical and isomeric SMILES for each compound in cidsHit. Load the downloaded data into a new data frame called dfRaw. Print the size (or dimension) of the data frame using Dimensions.
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 5. Create a new data frame called df, which combines the data stored in cidsHit and dfRaw.
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 6. Remove from df the compounds that violate any criterion of Congreve's rule of 3 and show the number of remaining compounds (the number of rows of df).
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 7: Get the unique canonical SMILES strings from the df.
In[]:=
(*Writeyourcodeinthiscell*)
Exercise 8: identify the top 10 compounds that were returned from the largest number of query compounds among those with unique canonical SMILEs strings.
◼
Sort the data frame by the number of times returned (in descending order) and then by CID (in ascending order)
◼
For each of the 10 compounds, print its CID, isomeric SMILES, and the number of times it was returned.
◼
For each of the 10 compounds, draw its structure (using isomeric SMILES).
(these can be separate steps, saving the results at each step)
In[]:=
(*Writeyourcodeinthiscell*)
Attributions
Attributions
Adapted from the corresponding OLCC 2019 Python Assignment:
https://chem.libretexts.org/Courses/Intercollegiate_Courses/Cheminformatics_OLCC_ (2019)/7%3 A__Computer-Aided_Drug _Discovery _and _Design/7.2%3 A_Python _Assignment-Virtual_Screening
https://chem.libretexts.org/Courses/Intercollegiate_Courses/Cheminformatics_OLCC_ (2019)/7%3 A__Computer-Aided_Drug _Discovery _and _Design/7.2%3 A_Python _Assignment-Virtual_Screening
Cite this as: Joshua Schrier, "07 Virtual Screening" from the Notebook Archive (2020), https://notebookarchive.org/2020-10-ebo06m8
Download
